Dendrit
Новости, статьи, учебные материалы по медицине
ПОДПИСАТЬСЯ НА РАССЫЛКУ
Ответы 61-80
61. Биосинтез мочевины. Связь орнитинового цикла с превращениями фумаровой и аспарагиновой кислот. Причины гипераммониемии. Уремия как следствие нарушения выведения мочевины из организма.
Биосинтез мочевины – основной путь обезвреживания аммиака. Мочевина синтезируется в орнитиновом цикле, протекающем в клетках печени. Эту последовательность реакций открыли Х.Кребс и К.Хензелейт в 1932 г. Согласно современным представлениям, цикл мочевины включает последовательность пяти реакций.
Две начальные реакции биосинтеза мочевины происходят в митохондриях клеток печени.

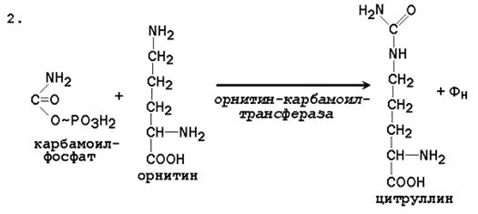
Последующие реакции протекают в цитоплазме клеток печени.
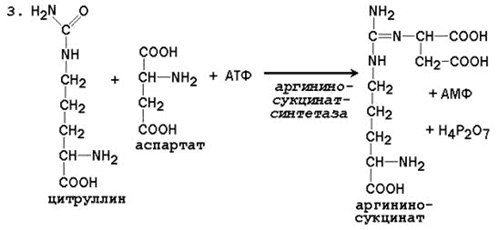
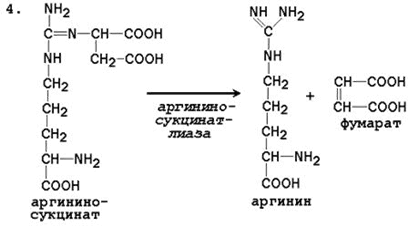
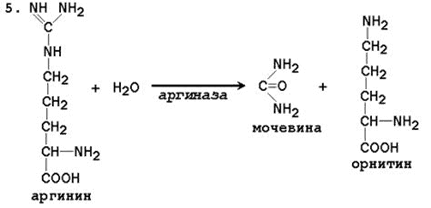
Общая схема орнитинового цикла представлена на рисунке 24.2:
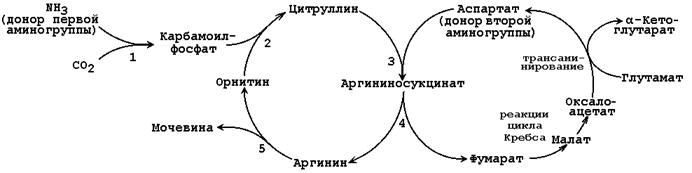
Рисунок 24.2. Схема орнитинового цикла и его связь с превращениями фумаровой и аспарагиновой кислот.
Цифрами обозначены ферменты, катализирующие реакции орнитинового цикла: 1 – карбамоилфосфатсинтетаза; 2 – орнитин-карбамоилтрансфераза; 3 – аргининосукцинатсинтетаза; 4 – аргининосукцинатлиаза; 5 – аргиназа.
24.4.2. Орнитиновый цикл находится в тесной взаимосвязи с циклом трикарбоновых кислот:
- пусковые реакции цикла мочевины, как и реакции ЦТК, протекают в митохондриальном матриксе;
- поступление СО2 и АТФ, необходимых для образования мочевины, обеспечивается работой ЦТК;
- в цикле мочевины образуется фумарат, который является одним из субстратов ЦТК. Фумарат гидратируется в малат, который в свою очередь окисляется в оксалоацетат. Оксалоацетат может подвергаться трансаминированию в аспартат; эта аминокислота участвует в образовании аргининосукцината.
24.4.3. Регуляция активности ферментов цикла осуществляется главным образом на уровне карбамоилфосфатсинтетазы, которая малоактивна в отсутствие своего аллостерического активатора - N-ацетил-глутамата. Концентрация последнего зависит от концентрации его предшественников (ацетил-КоА и глутамата), а также аргинина, который является аллостерическим активатором N-ацетилглутаматсинтазы:
Ацетил-КоА + Глутамат N-ацетилглутамат + КоА-SH
Концентрация ферментов орнитинового цикла зависит от содержания белка в пищевом рационе. При переходе на диету, богатую белком, в печени повышается синтез ферментов орнитинового цикла. При возвращении к сбалансированному рациону концентрация ферментов снижается. В условиях голодания, когда усиливается распад тканевых белков и использование аминокислот как энергетических субстратов, возрастает продукция аммиака, концентрация ферментов орнитинового цикла увеличивается.
24.4.4. Нарушения орнитинового цикла. Известны метаболические нарушения, обусловленные частичным блокированием каждого из 5 ферментов, катализирующих в печени реакции синтеза мочевины, а также N-ацетилглутаматсинтазы. Эти генетические дефекты, очевидно, являются частичными. Полное блокирование какой-либо из стадий цикла мочевины в печени, по-видимому, несовместимо с жизнью, потому что другого эффективного пути удаления аммиака не существует.
Общим признаком всех нарушений синтеза мочевины является повышенное содержание NH4+ в крови (гипераммониемия). Наиболее тяжёлые клинические проявления наблюдаются при дефекте фермента карбамоилфосфатсинтетазы. Клиническими симптомами, общими для всех нарушений цикла мочевины, являются рвота, нарушение координации движений, раздражительность, сонливость и умственная отсталость. Если заболевание не диагностируется, то быстро наступает гибель. У детей старшего возраста проявлениями заболевания служат повышенная возбудимость, увеличение размеров печени и отвращение к пище с высоким содержанием белка.
Лабораторная диагностика заболеваний включает определение содержания аммиака и метаболитов орнитинового цикла в крови, моче и спинномозговой жидкости; в сложных случаях прибегают к биопсии печени.
Значительное улучшение наблюдается при ограничении белка в диете, при этом могут быть предотвращены многие нарушения мозговой деятельности. Малобелковая диета приводит к снижению содержания аммиака в крови и к улучшению клинической картины при мягких формах этих наследственных нарушений. Пищу следует принимать часто, небольшими порциями, для того чтобы избежать резкого повышения уровня аммиака в крови.
24.4.5. Клинико-диагностическое значение определения мочевины в крови и моче. В крови здорового человека содержание мочевины составляет 3,33 – 8,32 ммоль/л. За сутки с мочой выводится 20 – 35 г мочевины.
Изменения содержания мочевины в крови при заболеваниях зависят от соотношения процессов её образования в печени и выведения почками. Повышение содержания мочевины в крови (гиперазотемия) отмечается при почечной недостаточности, снижение – при недостаточности печени, при диете с низким содержанием белков.
Повышение экскреции мочевины с мочой наблюдается при употреблении пищи с высоким содержанием белков, при заболеваниях, сопровождающихся усилением катаболизма белков в тканях, при приёме некоторых лекарств (например, салицилатов). Снижение экскреции мочевины с мочой характерно для заболеваний и токсических поражений печени, заболеваний почек, сопровождающихся нарушением их фильтрационной способности.
62. Обмен глутамата и аспартата, роль в азотистом обмене, распад до конечных продуктов.
Аммиак, образующийся в тканях, сначала превращается в нетоксичное соединение и в таком виде переносится кровью к печени или почкам. Такими транспортными формами являются аминокислоты глутамин, аспарагин и аланин.
24.2.2. Образование глутамина и аспарагина из глутамата и аспартата соответственно происходит во многих тканях, включая головной мозг:
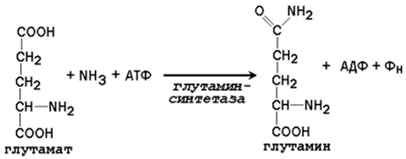
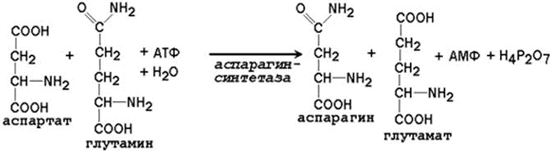
Глутамин - нейтральное нетоксичное соединение, способное легко проходить через клеточные мембраны. В виде этой аминокислоты аммиак транспортируется в крови. В крови здоровых людей содержание глутамина существенно превышает содержание других аминокислот. Глутамин, помимо участия в синтезе белка, служит источником азота в биосинтезе гистидина, глюкозамина, пуриновых и пиримидиновых нуклеотидов. С кровью глутамин поступает в печень и почки. Здесь он под действием фермента глутаминазы превращается в глутамат и аммиак. При участии аспарагиназы также происходит образование аммиака из аспарагина.
24.2.3. Аланин является транспортной формой аммиака, которая образуется преимущественно в мышцах. При интенсивной физической нагрузке источниками аммиака служат реакции дезаминирования аминокислот и аденозинмонофосфата (АМФ). Сначала аммиак превращается в аминогруппу глутамата в реакции восстановительного аминирования , катализируемой глутаматдегидрогеназой(см. параграф 18.6.2):
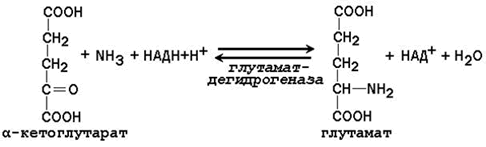
Образовавшийся глутамат переносит затем свою α-аминогруппу на пируват, всегда имеющийся в достаточном количестве, поскольку это продукт протекающего в мышцах гликолиза. Реакция катализируется аланинаминотрансферазой.
Глутамат + Пируват α-Кетоглутарат + Аланин
Аланин (нейтральная аминокислота, не несущая суммарного заряда при значениях рН, близких к 7) выходит из клеток и доставляется кровью к печени. Здесь он под действием аланинаминотрансферазы передаёт свою аминогруппу α-кетоглутарату, в результате чего образуется глутамат.
α-Кетоглутарат + Аланин Глутамат + Пируват
Далее в реакции, катализируемой глутаматдегидрогеназой, глутамат дезаминируется с образованием α-кетоглутарата и аммиака, который в печени превращается в мочевину.
63. Роль серина и глицина в образовании одноуглеродных групп и их использование в биологических синтезах. Участие ТГФК в этих процессах.
Главную роль в реакциях обмена серина и глицина играют ферменты, в состав которых в качестве кофермента входит тетрагидрофолиевая кислота (ТГФК). ТГФК образуется в организме в результате восстановления фолиевой кислоты (витамина Вс).
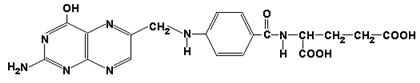
фолиевая кислота
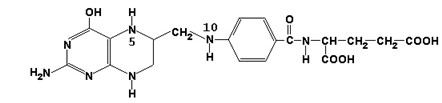
ТГФК
25.1.2. Реакционноспособными центрами в молекуле ТГФК являются атомы азота в положениях 5 и 10. Атомы водорода при N5 и N10 могут замещаться на различные одноуглеродные группы: метильную (-СН3), метиленовую (-СН2-), метенильную (=СН-), формильную (-СН=О) и некоторые другие. Основными источниками одноуглеродных групп в клетке служат серин и глицин.
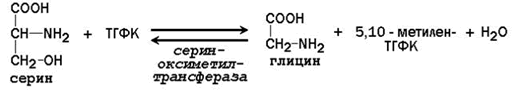

5,10-Метилен-ТГФК используется как донор метильной группы в реакциях биосинтеза тимидилового нуклеотида.
При окислении 5,10-метилен-ТГФК образуются 5,10-метенил-ТГФК и 10-формил-ТГФК. Эти производные ТГФК служат источниками атомов углерода в процессе биосинтеза пуриновых нуклеотидов (аденилового и гуанилового).
При восстановлении 5,10-метилен-ТГФК образуется 5-метил-ТГФК. Это соединение интересно тем, что может поставлять метильную группу для регенерации метионина из гомоцистеина (см. далее).
25.1.3. Аминокислота глицин, помимо участия в синтезе белка и образовании различных одноуглеродных групп, является предшественником ряда специализированных биомолекул:
- оба атома углерода и атом азота глицина могут включаться в структуру пуринового ядра (атомы С4, С5 и N7);
- глицин является главным предшественником порфиринов (простетической группы гемоглобина, миоглобина, цитохромов);
- глицин участвует в синтезе креатина - предшественника креатинфосфата, участвующего в биоэнергетике мышечной и нервной ткани;
- глицин входит в состав пептидного кофермента глутатиона;
- участвует в образовании конъюгатов (гликохолевая кислота, гиппуровая кислота).
64. Метионин и S-аденозилметионин: строение, участие в процессах трансметилирования. Регенерация S-аденозилметионина из гомоцистеина..
Метильная группа метионина, связанная с атомом серы, также представляет собой подвижную одноуглеродную группу, способную участвовать в реакциях трансметилирования (переноса метильной группы). Активной формой метионина, принимающей непосредственное участие в этих превращениях, является S-аденозилметионин, который образуется при взаимодействии метионина с АТФ.
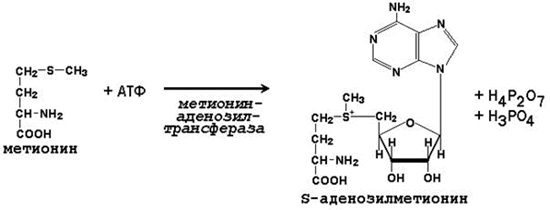
Примеры реакций трансметилирования с участием S-аденозилметионина приводятся в таблице 25.1.
Таблица 25.1
Использование метильной группы S-аденозилметионина в реакциях трансметилирования
| Субстрат | Метилированный продукт |
|---|---|
| Норадреналин | Адреналин |
| Адреналин | Метоксиадреналин |
| Гуанидинацетат | Креатин |
| Карнозин | Ансерин |
| Гистамин | N-метилгистамин |
| Фосфатидилэтаноламин | Фосфатидилхолин |
Вот некоторые примеры этих реакций.
1) Образование фосфатидилхолина из фосфатидилэтаноламина - ключевая реакция синтеза фосфолипидов:
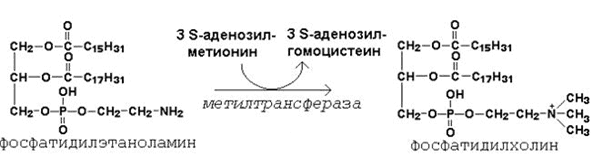
Фосфатидилхолин – главный фосфолипидный компонент биологических мембран; он входит в состав липопротеинов, принимает участие в транспорте холестерола и триацилглицеролов; нарушение синтеза фосфатидилхолина в печени приводит к жировой инфильтрации.
2) Образование адреналина из норадреналина - заключительная реакция синтеза гормона мозгового вещества надпочечников:
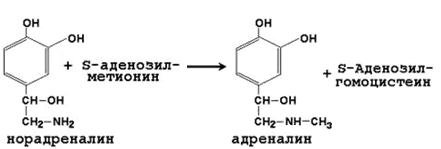
Адреналин выделяется в кровь при эмоциональном стрессе и участвует в регуляции углеводного и липидного обмена в организме.
3) Реакции метильной конъюгации - один из этапов обезвреживания чужеродных соединений и эндогенных биологически активных веществ:
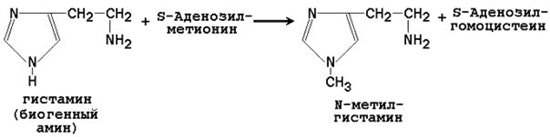
В результате метилирования блокируются реакционноспособные SH- и NН-группы субстратов. Продукты реакции не обладают активностью и выводится из организма с мочой.
25.2.3. После отдачи метильной группы S-аденозилметионин превращается в S-аденозилгомоцистеин. Последний расщепляется на аденозин и гомоцистеин. Гомоцистеин может вновь превращаться в метионин за счёт метильной группы 5-метил-ТГФК (см. предыдущий параграф):
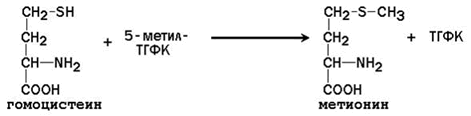
В этой реакции в качестве кофермента участвует метилкобаламин – производное витамина В12. При недостатке витамина В12 нарушается синтез метионина из гомоцистеина и накапливается 5-метил-ТГФК. Так как реакция образования 5-метил-ТГФК из 5,10-метилен-ТГФК необратима, одновременно возникает дефицит фолиевой кислоты.
25.2.4. Другим путём использования гомоцистеина, как уже упоминалось, является участие в синтезе цистеина. Биологическая роль цистеина:
- входит в состав белка, где может образовывать дисульфидные связи, стабилизирующие пространственную структуру макромолекулы;
- участвует в синтезе глутатиона, причём цистеиновая SH-группа определяет реакционную способность этого кофермента;
- является предшественником тиоэтаноламина в молекуле HS-КоА;
- служит предшественником таурина в конъюгированных желчных кислотах;
- является источником атома серы в органических сульфатах (хондроитинсульфат, гепарин, ФАФС).
65. Обмен фенилаланина и тирозина. Использование тирозина для синтеза катехоламинов, тироксина, меланинов. Распад тирозина до конечных продуктов. Наследственные нарушения обмена фенилаланина и тирозина (фенилкетонурия, алкаптонурия, альбинизм).
Обмен фенилаланина и тирозина в тканях человека можно представить в следующем виде (см. рисунок 25.1).
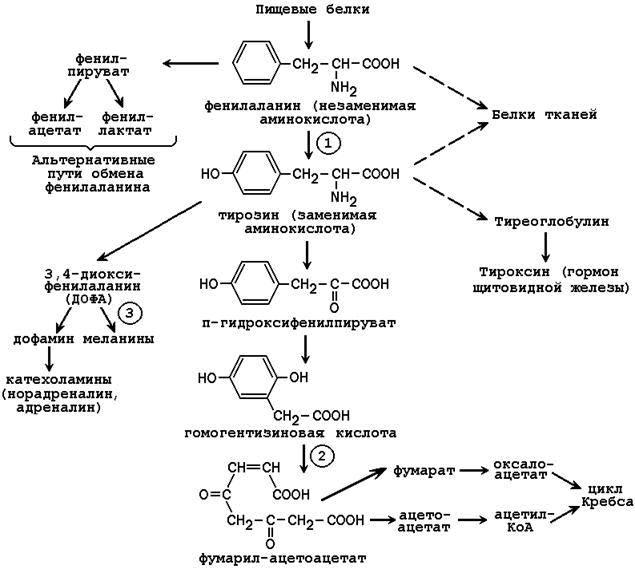
Рисунок 25.1. Пути обмена фенилаланина и тирозина в тканях (цифрами обозначены наиболее часто встречающиеся дефекты ферментов; далее приводится характеристика этих нарушений).
25.4.2. Известен ряд врождённых нарушений обмена фенилаланина и тирозина.
Фенилкетонурия – врождённое нарушение процесса гидроксилирования фенилаланина до тирозина. Заболевание чаще всего вызвано отсутствием или недостатком фермента фенилаланингидроксилазы (обозначен цифрой 1 на рисунке 25.1), реже - нарушением образования тетрагидробиоптерина.
Ранними симптомами фенилкетонурии являются повышенная возбудимость и двигательная активность, рвота и трудности вскармливания, с 3 – 5-го месяца нарушается интеллектуальное развитие, исчезает реакция на окружающее. Со временем у детей появляются судороги. Волосы и глаза обычно менее пигментированы, чем у других членов семьи. При отсутствии лечения продолжительность жизни больных составляет 20 - 30 лет.
Биохимическая основа фенилкетонурии – накопление фенилаланина в организме. Высокая концентрация аминокислоты стимулирует выработку фермента, превращающего фенилаланин вфенилпируват (в норме этот фермент малоактивен). Путём восстановления фенилпируват переходит в фениллактат, а путём декарбоксилирования – в фенилацетат. Эти продукты наряду с фенилаланином в существенных количествах обнаруживаются в моче больных.
В настоящее время имеются достоверные свидетельства того, что за токсическое повреждение мозга ответственны главным образом высокие концентрации фенилаланина. Повышенное содержание фенилаланина тормозит транспорт тирозина и других аминокислот через биологические мембраны. Это приводит к ограничению синтеза белка в клетках мозга и нарушению синтеза нейромедиаторов.
Раннюю диагностику заболевания нельзя провести исходя только из клинической симптоматики. Диагноз ставится биохимически путём скрининга всех новорождённых. Лечение больных фенилкетонурией основано на ограничении поступления фенилаланина в организм и снижения концентрации этой аминокислоты в плазме. С этой целью используются искусственные питательные смеси, в которых фенилаланин отсутствует (например, берлофен).
Алкаптонурия – врожденное нарушение обмена фенилаланина, вызванное отсутствием фермента оксидазы гомогентизиновой кислоты (цифра 2 на рисунке 25.1). Это приводит к нарушению образования малеилацетоацетата, расщепляющегося далее до фумарата и ацетоацетата. В раннем детском возрасте единственным проявлением дефицита фермента является изменение окраски мочи. Гомогентизиновая кислота секретируется в просвет канальцев и в значительном количестве выводится с мочой. На воздухе она окисляется, а затем полимеризуется в окрашенное соединение, которое окрашивает пелёнки в чёрный цвет. Экскреция гомогентизиновой кислоты зависит от содержания фенилаланина и тирозина в пище.
Следствием накопления гомогентизиновой кислоты в организме является охроноз - шиферно-голубой оттенок ушного и носового хрящей, вызванный накоплением в них пигмента. Развитие охроноза можно предотвратить, если с раннего возраста ограничивать поступление с пищей фенилаланина и тирозина.
Альбинизм развивается при отсутствии в пигментных клетках фермента тирозиназы (обозначена цифрой 3 на рисунке 25.1), которая участвует в образовании меланина. В результате волосы, кожа и глаза больного лишены этого пигмента. При альбинизме наблюдается повышение чувствительности к солнечным лучам и некоторое нарушения зрения.
66. Синтез гема и гемоглобина. Распад гемоглобина, обмен желчных пигментов. Нарушения обмена желчных пигментов. Значение определения желчных пигментов в диагностике желтух. Условно физиологическая желтуха новорожденных.
Хромопротеины относятся к сложным белкам. Молекулы хромопротеинов состоят из полипептидных цепей и небелковых компонентов (простетических групп), из которых наиболее распространённым является гем.
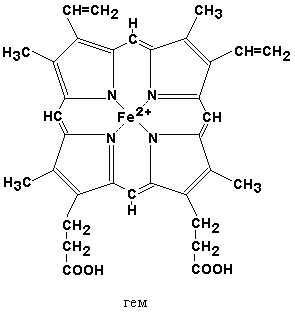
26.1.2. Гем в качестве простетической группы содержат следующие белки:
- Гемоглобин – присутствует в эритроцитах; в его состав входят 4 полипептидные цепи, с каждой из которых связана одна гемовая группа. Этот белок транспортирует О2 и СО2 в крови.
- Миоглобин – присутствует в клетках мышечной ткани; представляет собой одну полипептидную цепь, с которой связана одна гемовая группа. Этот белок запасает кислород в мышцах и отдаёт его при выполнении мышечной работы.
- Цитохромы – белки-ферменты, содержатся в митохондриях клеток, участвуют в переносе электронов на кислород в дыхательной цепи.
- Пероксидаза и каталаза – белки-ферменты, ускоряют расщепление пероксида водорода Н2О2 на Н2О и О2.
Схема биосинтеза гемоглобина представлена на рисунке 26.1. Исходными веществами в этом метаболическом пути являются аминокислота глицин и метаболит цикла Кребсасукцинил-КоА. Синтез происходит в ретикулоцитах (незрелых эритроцитах, содержащих клеточное ядро). Реакции идут в митохондриях и цитоплазме клеток.
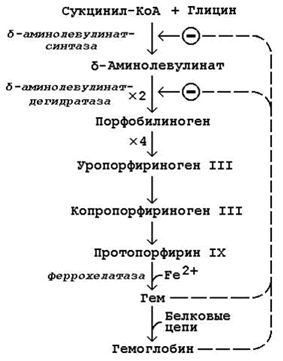
Рисунок 26.1. Биосинтез гемоглобина и его регуляция.
Первая стадия в последовательности реакций, ведущих к синтезу гема, катализируется δ-аминолевулинат-синтазой. Фермент абсолютно специфичен к субстратам; кофакторами фермента являются пиридоксаль-5-фосфат и ионы Mg2+.
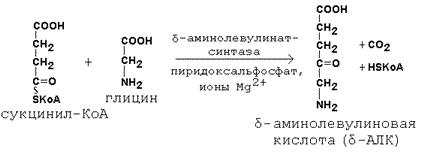
Имеются данные о том, что некоторые лекарственные препараты, а также стероидные гормоны, напротив, индуцируют синтез печёночной δ-аминолевулинат-синтазы.
Во второй реакции, катализируемой δ-аминолевулинат-дегидратазой, при конденсации двух молекул δ-аминолевулината образуется порфобилиноген.
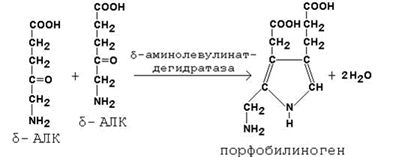
В дальнейшем из четырёх молекул порфобилиногена в результате ряда сложных ферментативных реакций образуется протопорфирин IX – непосредственный предшественник гема. При участии митохондриального фермента феррохелатазы двухвалентное железо включается в уже готовую структуру протопорфирина. Для протекания этой реакции необходимы аскорбиновая кислота и цистеин в качестве восстановителей. Ингибитором феррохелатазы является свинец. На заключительном этапе происходит соединение гема с белковыми цепями, характерными для синтезируемого хромопротеина. Конечные продукты этого биосинтеза (гем, гемоглобин) подавляют начальные реакции по механизму отрицательной обратной связи (рисунок 9).
При врождённых и приобретённых нарушениях биосинтеза гема развиваются заболевания – порфирии.
26.2.2. Порфирии – группа наследственных заболеваний, обусловленных частичным дефицитом одного из ферментов синтеза гема. Снижение образования гема приводит к снятию его ингибирующего эффекта на начальные этапы биосинтеза, результатом чего является избыточное образование порфиринов и их предшественников. Основными симптомами порфирий являются:
- нарушения со стороны центральной нервной системы (т.к. предшественники порфиринов являются нейротоксинами);
- повышенная светочувствительность кожи (порфирины накапливаются в коже, поглощают свет и переходят в возбуждённое состояние, вызывая образование токсичных свободных радикалов);
- анемия (снижение содержания гемоглобина в крови) ;
- порфиринурия - выведение порфиринов с мочой и калом (моча приобретает красную окраску).
Порфиринурия может также развиваться при отравлениях свинцом.
Содержание гемоглобина в крови здоровых людей составляет 130-160 г/л. Гемоглобин крови полностью обновляется в течение 120 дней (продолжительность жизни эритроцита).
Разрушение эритроцитов и начальные этапы катаболизма гема происходят в клетках ретикуло-эндотелиальной системы (РЭС), которые находятся в печени (клетки Купфера), селезёнке, костном мозге. Схема катаболизма гемоглобина в тканях приводится на рисунке 26.3.
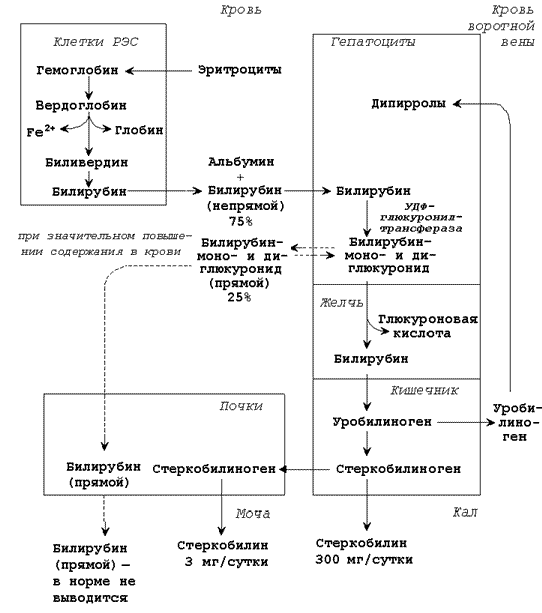
Рисунок 26.3. Схема катаболизма гемоглобина в тканях.
26.4.2. Продукты распада гема называют желчными пигментами, так как все они в разных количествах обнаруживаются в желчи. К желчным пигментам относятся: биливердин (зелёного цвета), билирубин (красно-коричневого цвета), уробилиноген и стеркобилиноген (бесцветные), уробилин и стеркобилин (жёлтого цвета). Далее приводятся формулы билирубина и его диглюкуронида.
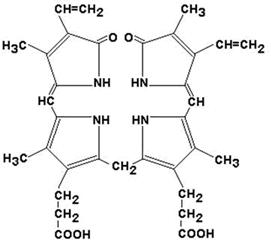 |
Билирубин (свободный или неконъюгированный билирубин) образуется в клетках ретикуло-эндотелиальной системы (РЭС), транспортируется в гепатоциты. Билирубин нерастворим в воде и растворим в жирах, токсичен, в крови присутствует в виде комплекса с альбумином, не проникает через почечный фильтр. Эта фракция билирубина в плазме крови называется непрямым билирубином, так как взаимодействует с диазореактивом только после осаждения альбуминов. |
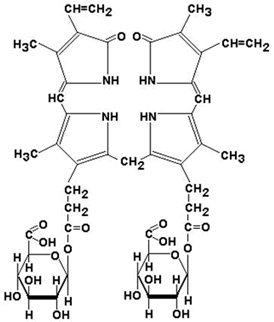 |
Билирубиндиглюкуронид (связанный или конъюгированный билирубин) образуется в гепатоцитах под действием фермента билирубин-глюкуронилтрансферазы, путём активного транспорта выводится в желчные канальцы. Он хорошо растворим в воде и не растворим в жирах, малотоксичен, в крови не связан с белками плазмы, может проникать через почечный фильтр. Эта фракция билирубина в плазме крови называется прямым билирубином, так как непосредственно может взаимодействовать с диазореактивом. |
Общее содержание билирубина в крови здорового человека составляет 8 – 20 мкмоль/л, из них 6 – 15 мкмоль/л приходится на непрямой билирубин, 2 – 5 мкмоль/л – на прямой билирубин. Увеличение общего билирубина в крови (более 27 мкмоль/л) приводит к окрашиванию кожи, слизистых оболочек, склеры глаз в жёлтый цвет (желтуха). Определение содержания желчных пигментов в крови используют при выяснении происхождения желтух. Желтуха бывает надпечёночная (гемолитическая), печёночная (паренхиматозная), подпечёночная (обтурационная или механическая).
26.5.2. Надпечёночная (гемолитическая) желтуха вызвана массивным распадом эритроцитов в результате резус-конфликта, попадания в кровь веществ, вызывающих разрушение мембран эритроцитов и некоторых других заболеваниях. При этой форме желтухи в крови повышено содержание непрямого билирубина, в моче повышено содержание стеркобилина, билирубин отсутствует, в кале повышено содержание стеркобилина.
26.5.3. Печёночная (паренхиматозная) желтуха вызвана повреждением клеток печени при инфекциях и интоксикациях. При этой форме желтухи в крови повышено содержание непрямого и прямого билирубина, в моче повышено содержание уробилина, присутствует билирубин, в кале понижено содержание стеркобилина.
26.5.4. Подпечёночная (обтурационная) желтуха вызвана нарушением оттока желчи, например, при закупорке желчевыводящего протока камнем. При этой форме желтухи в крови повышено содержание прямого билирубина (иногда и непрямого), в моче отсутствует стеркобилин, присутствует билирубин, в кале понижено содержание стеркобилина.
26.5.5. Условно физиологическая желтуха новорождённых развивается у большинства здоровых новорождённых в первые дни после рождения и продолжается около двух недель. При различных заболеваниях, возникающих у новорождённых, а также у недоношенных детей желтушный период затягивается. Увеличение длительности гипербилирубинемии может привести к серьёзным последствиям: накоплению билирубина в ткани мозга (ядерная желтуха).
Повышению содержания билирубина в крови новорождённых могут способствовать следующие особенности обмена веществ в их организме:
- замена фетального гемоглобина на гемоглобин А. В первые дни после рождения усиливается гемолиз эритроцитов, содержващих HbF; образуются новые эритроциты, содержащие HbA. HbF подвергается катаболизму; образуется значительное количество билирубина;
- отвлечение альбуминов плазмы для транспорта жирных кислот. Содержание углеводов в организме новорождённых сравнительно невелико; основным энергетическим субстратом являются жирные кислоты, концентрация которых в крови повышается, жирные кислоты транспортируются в комплексе с альбуминами;
- низкая активность глюкуронилтрансферазы в ткани печени. Замедление процессов конъюгации билирубина в печени затрудняет его выведение в кишечник;
- стерильность кишечника. В кишечнике новорождённого отсутствует микрофлора, поэтому билирубин не превращается в стеркобилиноген и может происходить его обратное всасывание в кровоток.
67. Обмен железа. Суточная потребность, источники, всасывание, транспорт, депонирование, использование в организме, реутилизация железа.
В организме человека содержится 4 – 6 г железа. Из этого количества 65-70% приходится на долю гемоглобина. Значительно меньше Fе находится в составе других гемсодержащих белков (миоглобин, цитохромы), а также металлопротеинов (ферритин, трансферрин). Поэтому обмен железа в организме определяется прежде всего синтезом и распадом гемоглобина эритроцитов. Недостаточное поступление железа в организм проявляется в первую очередь как анемия (железодефицитная). Общая схема обмена железа представлена на рисунке 26.2.
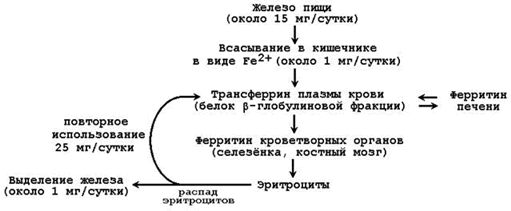
Рисунок 26.2. Обмен железа в организме.
26.3.2. В кишечнике всасывается лишь небольшая часть (около 1/10) имеющегося в пище железа. Транспортной формой железа в крови служит белок плазмы крови трансферрин. Другой белок, участвующий в метаболизме железа – ферритин – служит для депонирования железа, присутствует в большинстве тканей. Железо, освобождающееся при разрушении эритроцитов, может, как правило, повторно использоваться (реутилизироваться) для построения новых молекул хромопротеинов. Однако часть железа теряется организмом, главным образом, с желчью. Эти потери компенсируются поступлением железа с пищей.
68. Биосинтез пуриновых нуклеотидов. Происхождение атомов N и С пуринового кольца. Резервные пути биосинтеза пуриновых нуклеотидов. Распад пуриновых нуклеотидов. Особенности экскреции мочевой кислоты с мочой у детей раннего возраста. Нарушения обмена пуринов.
Ключевым соединением в биосинтезе как пуриновых, так и пиримидиновых нуклеотидов является 5-фосфорибозил-1-пирофосфат (ФРПФ). Это соединение участвует также в синтезе коферментов НАД+ и НАДФ+.
ФРПФ образуется при взаимодействии рибозо-5-фосфата и АТФ. Источниками рибозофосфата служат пентозофосфатный путь и распад нуклеотидов. Катализирует реакцию фермент ФРПФ-синтаза.
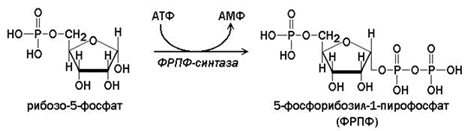
Внутриклеточная концентрация ФРПФ обычно низкая и строго регулируется. Скорость синтеза ФРПФ зависит от наличия субстратов синтеза, особенно рибозо-5-фосфата, и каталитической активности ФРПФ-синтазы, на которую влияют концентрация неорганического фосфата и концентрация АМФ, ГМФ и ИМФ, выступающих в качестве эффекторов.
26.8.2. Молекула ФРПФ служит основой для последующего синтеза пуринового ядра. Источниками атомов углерода и азота являются аминокислоты глутамин, глицин и аспартат, СО2 и два одноуглеродных производных ТГФК – формил-ТГФК и метенил-ТГФК (рисунок 26.7).
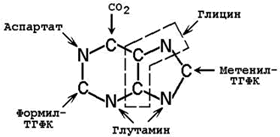
Рисунок 26.7. Происхождение атомов пуринового ядра.
Сначала в реакции, катализируемой фосфорибозил-пирофосфат-амидотрансферазой, из ФРПФ при участии глутамина образуется 5-фосфорибозиламин.
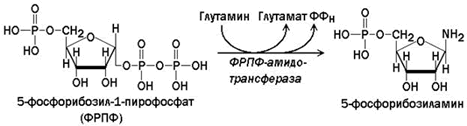
ФРПФ-амидотрансфераза – второй регуляторный фермент синтеза пуриновых нуклеотидов, он ингибируется АМФ и ГМФ по принципу обратной связи. Роль этого фермента в биосинтезе пуринов de novo , однако, менее существенна, чем ФРПФ-синтазы.
Далее к атому азота последовательно присоединяются все остальные компоненты пуринового ядра. Первым продуктом биосинтеза, содержащим готовую пуриновую структуру, является инозинмонофосфат (ИМФ). В его состав входит азотистое основание гипоксантин.
26.8.3. ИМФ является предшественником аденилового и гуанилового нуклеотидов (рисунок 26.5). В синтезе АМФ из ИМФ при взаимодействии аспартатом образуется аденилосукцинат. В следующей реакции отщепляется фумарат и образуется АМФ.
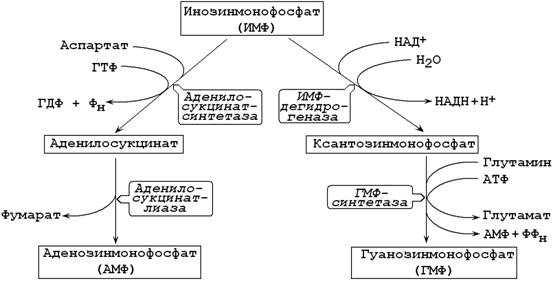
Рисунок 26.8. Образование АМФ и ГМФ из инозинмонофосфата.
Синтез ГМФ из ИМФ также включает две стадии. Сначала ИМФ окисляется в ксантозинмонофосфат, затем добавляется NH2-группа из глутамина.
Интересно отметить, что синтез АМФ требует участия ГТФ, а синтез ГМФ – участия АТФ. Эта особенность биосинтеза способствует поддержанию нужного соотношения адениловых и гуаниловых нуклеотидов в клетке.
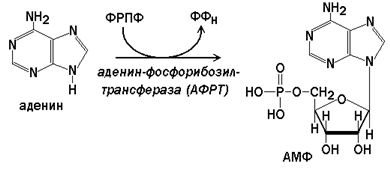
26.8.4. Наряду с биосинтезом пуриновых нуклеотидов в клетке de novo существуют пути регенерации пуриновых нуклеотидов из свободных азотистых оснований, образующихся при гидролизе нуклеиновых кислот и нуклеотидов. Эти реакции проще, чем пути синтеза нуклеотидов de novo, и энергетическая цена их значительно меньше. Наибольшее значение имеет механизм фосфорибозилирования пуриновых оснований.
В клетках имеются 2 фермента, участвующих в реакциях синтеза нуклеотидов из пуриновых оснований.
Аденин-фосфорибозилтрансфераза (АФРТ) катализирует перенос фосфорибозы с ФРПФ на аденин:
Гипоксантин-гуанин-фосфорибозилтрансфераза(ГГФРТ) катализирует перенос фосфорибозы с ФРПФ на гуанин или гипоксантин:
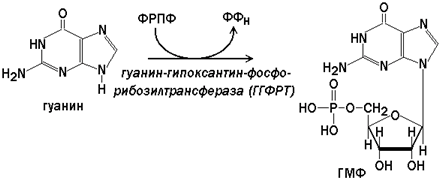
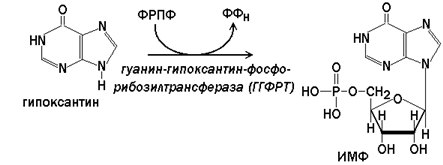
Реакции с участием второго фермента протекают более активно, чем синтез АМФ из аденина.
26.8.5. Нарушения обмена пуриновых нуклеотидов. При нарушениях пуринового обмена часто наблюдается гиперурикемия – повышение содержания мочевой кислоты в крови. Гиперурикемия может быть первичной или вторичной.
Первичная гиперурикемия является ведущим симптомом подагры – полиэтиологического заболевания, как правило, наследственной природы. Гиперурикемия при подагре обусловлена главным образом, избыточным образованием образованием мочевой кислоты, а также снижением её экскреции с мочой. Значительная и длительная гиперурикемия сопровождается отложением солей мочевой кислоты в хрящевой ткани, сухожилиях и слизистых сумках суставов. Накопление кристаллов уратов в тканях может вызывать резкую воспалительную реакцию (подагрический артрит), что приводит впоследствии к деформации сустава. Избыток мочевой кислоты способствует также образованию уратных камней в нижних отделах мочевыводящих путей.
Повышение уровня мочевой кислоты в крови отмечается также при наследственных дефектах некоторых ферментов:
Синдром Леша-Нихана (полное отсутствие ГГФРТ) наследуется как сцепленный с Х-хромосомой рецессивный признак. Болезнь характеризуется параличом, сопровождающимся судорогами, стремлением к членовредительству и тяжёлой гиперурикемией. Вследствие ферментативного дефекта нарушается переход гуанина и гипоксантина в ГМФ и ИМФ соответственно и указанные пуриновые основания превращаются в мочевую кислоту. Кроме того, повышенная концентрация ФРПФ способствует усилению синтеза пуринов de novo. Биохимическая основа неврологических отклонений при синдроме Леша-Нихана неизвестна.
Гликогеноз I типа или болезнь Гирке (дефицит глюкозо-6-фосфатазы) сопровождается повышением активности пентозофосфатного пути и приводит к повышению внутриклеточного уровня рибозо-5-фосфата, из которого синтезируется ФРПФ. Повышенный уровень ФРПФ приводит к увеличению синтеза пуринов de novo. Для данного заболевания характерен также лактатный ацидоз, приводящий к повышению порога секреции уратов почками; это способствует накоплению уратов в организме.
Вторичная гиперурикемия сопутствует заболеваниям, сопровождающимся усиленным распадом клеток (лейкозы, серповидно-клеточная анемия, сахарный диабет, псориаз).
Реже встречается гипоурикемия – снижение содержания мочевой кислоты в крови. Она может быть связана с понижением реабсорбции уратов из клубочкового фильтрата в почках. В этом случае наблюдается увеличение экскреции мочевой кислоты с мочой.
Гипоурикемия развивается и при недостаточности ксантиноксидазы, возникающей при генетическом дефекте фермента или при тяжёлом поражении печени. Это состояние сопровождается повышенной экскрецией гипоксантина и ксантина (ксантинурией), а также образованием в почках ксантиновых камней.
69. Регуляция метаболизма. Иерархия регуляторных систем. Значение эндокринной системы. Роль гормонов гипоталамуса и гипофиза.
Выучите определение понятия: гормоны – биологически активные соединения, выделяемые железами внутренней секреции в кровь или лимфу и оказывающие влияние на метаболизм клетки.
23.1.2. Запомните основные особенности действия гормонов на органы и ткани:
- гормоны синтезируются и выделяются в кровь специализированными эндокринными клетками;
- гормоны обладают высокой биологической активностью - физиологическое действие проявляется при концентрации их в крови порядка 10-6 - 10-12 моль/л;
- каждый гормон характеризуется присущей только ему структурой, местом синтеза и функцией; дефицит одного гормона не может быть восполнен другими веществами;
- гормоны, как правило, влияют на отдалённые от места их синтеза органы и ткани.
23.1.3. Гормоны осуществляют своё биологическое действие, образуя комплекс со специфическими молекулами – рецепторами. Клетки, содержащие рецепторы к определённому гормону, называются клетками-мишенями для этого гормона. Большинство гормонов взаимодействуют с рецепторами, расположенными на плазматической мембране клеток-мишеней; другие гормоны взаимодействуют с рецепторами, локализованными в цитоплазме и ядре клеток-мишеней. Имейте в виду, что дефицит как гормонов, так и их рецепторов может приводить к развитию заболеваний.
апомните, что в организме существует несколько уровней регуляции гомеостаза, которые тесно взаимосвязаны и функционируют как единая система (см. рисунок 23.1).
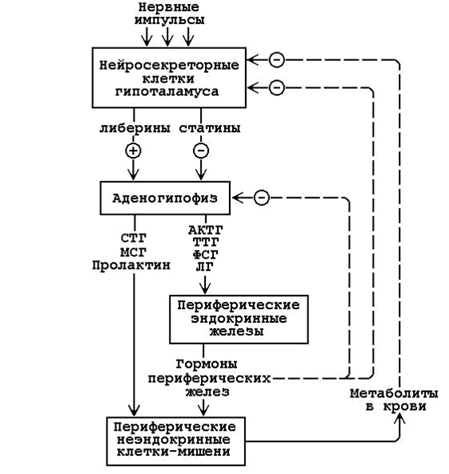
Рисунок 23.1. Иерархия регуляторных систем организма (пояснения в тексте).
23.2.2. 1. Сигналы из внешней и внутренней среды поступают в центральную нервную систему (высший уровень регуляции, осуществляет контроль в пределах целого организма). Эти сигналы трансформируются в нервные импульсы, попадающие на нейросекреторные клетки гипоталамуса. В гипоталамусе образуются:
- либерины (или рилизинг-факторы), стимулирующие секрецию гормонов гипофиза;
- статины – вещества, угнетающие секрецию этих гормонов.
Либерины и статины по системе портальных капилляров достигают гипофиза, где вырабатываются тропные гормоны. Тропные гормоны действуют на периферические ткани-мишени и стимулируют(знак “+”) образование и секрецию гормонов периферических эндокринных желёз. Гормоны периферических желёз угнетают (знак “–”) образование тропных гормонов, действуя на клетки гипофиза или нейросекреторные клетки гипоталамуса. Кроме того, гормоны, действуя на обмен веществ в тканях, вызывают изменения содержания метаболитов в крови, а те, в свою очередь, влияют (по механизму обратной связи) на секрецию гормонов в периферических железах (или непосредственно, или через гипофиз и гипоталамус).
2. Гипоталамус, гипофиз и периферические железы образуют средний уровень регуляции гомеостаза, обеспечивающий контроль нескольких метаболических путей в пределах одного органа, или ткани, или разных органов.
Гормоны эндокринных желёз могут влиять на обмен веществ:
- путём изменения количества ферментного белка;
- путём химической модификации ферментного белка с изменением его активности, а также
- путём изменения скорости транспорта веществ через биологические мембраны.
3. Внутриклеточные механизмы регуляции представляют собой низший уровень регуляции. Сигналами для изменения состояния клетки служат вещества, образующиеся в самих клетках или поступающие в неё.
Как уже упоминалось, местом непосредственного взаимодействия высших отделов центральной нервной системы и эндокринной системы является гипоталамус. Это небольшой участок переднего мозга, который расположен непосредственно над гипофизом и связан с ним при помощи системы кровеносных сосудов, образующих портальную систему.
23.4.1. Гормоны гипоталамуса. В настоящее время известно, что нейросекреторные клетки гипоталамуса продуцируют 7 либеринов (соматолиберин, кортиколиберин, тиреолиберин, люлиберин, фоллиберин, пролактолиберин, меланолиберин) и 3 статина (соматостатин, пролактостатин, меланостатин). Все эти соединения являются пептидами.
Гормоны гипоталамуса через специальную портальную систему сосудов попадают в переднюю долю гипофиза (аденогипофиз). Либерины стимулируют, а статины подавляют синтез и секрецию тропных гормонов гипофиза. Эффект либеринов и статинов на клетки гипофиза опосредуется цАМФ- и Са2+-зависимыми механизмами.
Характеристика наиболее изученных либеринов и статинов приведена в таблице 23.2.
| Фактор | Место действия | Основные биологические эффекты | Регуляция секреции |
|---|---|---|---|
| Кортиколиберин | Аденогипофиз | Стимулирует секрецию адренокортикотропного гормона (АКТГ) | Секреция стимулируется при стрессах и подавляется АКТГ |
| Тиреолиберин | - “ – “ - | Стимулирует секрецию тиреотропного гормона (ТТГ) и пролактина | Секрецию тормозят тиреоидные гормоны |
| Соматолиберин | - “ – “ - | Стимулирует секрецию соматотропного гормона (СТГ) | Секрецию стимулирует гипогликемия |
| Люлиберин | - “ – “ - | Стимулирует секрецию фолликулостимулирующего гормона (ФСГ) и лютеинизирующего гормона (ЛГ) | У мужчин секреция вызывается снижением содержания тестостерона в крови, у женщин – снижением концентрации эстрогенов. Высокая концентрация ЛГ и ФСГ в крови подавляет секрецию |
| Соматостатин | - “ – “ - | Тормозит секрецию СТГ и ТТГ | Секреция вызывается физической нагрузкой. Фактор быстро инактивируется в тканях тела. |
| Пролактостатин | - “ – “ - | Тормозит секрецию пролактина | Секрецию стимулирует высокая концентрация пролактина и подавляют эстрогены, тестостерон и нервные сигналы при сосании. |
| Меланостатин | - “ – “ - | Угнетает секрецию МСГ (меланоцитостимулирующего гормона) | Секрецию стимулирует меланотонин |
23.4.2. Гормоны аденогипофиза. Аденогипофиз (передняя доля гипофиза) продуцирует и выделяет в кровь ряд тропных гормонов, регулирующих функцию как эндокринных, так и неэндокринных органов. Все гормоны гипофиза являются белками или пептидами. Внутриклеточным посредником всех гипофизарных гормонов (кроме соматотропина и пролактина) служит циклический АМФ (цАМФ). Характеристика гормонов передней доли гипофиза приводится в таблице 3.
| Гормон | Ткань-мишень | Основные биологические эффекты | Регуляция секреции |
|---|---|---|---|
| Адренокортикотропный гормон (АКТГ) | Кора надпочечников | Стимулирует синтез и секрецию стероидов корой надпочечников | Стимулируется кортиколиберином |
| Тиреотропный гормон (ТТГ) | Щитовидная железа | Усиливает синтез и секрецию тиреоидных гормонов | Стимулируется тиреолиберином и подавляется тиреоидными гормонами |
| Соматотропный гормон (гормон роста, СТГ) | Все ткани | Стимулирует синтез РНК и белка, рост тканей, транспорт глюкозы и аминокислот в клетки, липолиз | Стимулируется соматолиберином, подавляется соматостатином |
| Фолликулостимулирующий гормон (ФСГ) | Семенные канальцы у мужчин, фолликулы яичников у женщин | У мужчин повышает образование спермы, у женщин – образование фолликулов | Стимулируется люлиберином |
| Лютеинизирующий гормон (ЛГ) | Интерстициальные клетки семенников (у мужчин) и яичников (у женщин) | Вызывает секрецию эстрогенов, прогестерона у женщин, усиливает синтез и секрецию андрогенов у мужчин | Стимулируется люлиберином |
| Пролактин | Молочные железы (альвеолярные клетки) | Стимулирует синтез белков молока и развитие молочных желёз | Подавляется пролактостатином |
| Меланоцитостимулирующий гормон (МСГ) | Пигментные клетки | Повышает синтез меланина в меланоцитах (вызывает потемнение кожи) | Подавляется меланостатином |
23.4.3. Гормоны нейрогипофиза. К гормонам, секретируемым в кровоток задней долей гипофиза, относятся окситоцин и вазопрессин. Оба гормона синтезируются в гипоталамусе в виде белков-предшественников и перемещаются по нервным волокнам в заднюю долю гипофиза.
Окситоцин – нонапептид, вызывающий сокращения гладкой мускулатуры матки. Он используется в акушерстве для стимуляции родовой деятельности и лактации.
Вазопрессин – нонапептид, выделяемый в ответ на повышение осмотического давления крови. Клетками-мишенями для вазопрессина являются клетки почечных канальцев и гладкомышечные клетки сосудов. Действие гормона опосредовано цАМФ. Вазопрессин вызывает сужение сосудов и повышение артериального давления, а также усиливает реабсорбцию воды в почечных канальцах, что приводит к снижению диуреза.
23.4.4. Основные виды нарушений гормональной функции гипофиза и гипоталамуса. При дефиците соматотропного гормона, возникающем в детском возрасте, развивается карликовость (низкий рост). При избытке соматотропного гормона, возникающем в детском возрасте, развивается гигантизм (аномально высокий рост).
При избытке соматотропного гормона, возникающем у взрослых (в результате опухоли гипофиза), развивается акромегалия – усиленный рост кистей рук, ступней, нижней челюсти, носа.
При недостатке вазопрессина, возникающем вследствие нейротропных инфекций, черепно-мозговых травм, опухолей гипоталамуса, развивается несахарный диабет. Основным симптомом этого заболевания является полиурия – резкое увеличение диуреза при пониженной (1,001 – 1,005) относительной плотности мочи.
70. Механизм действия дистантных гормонов. Роль мембраносвязанных ферментов в передаче внешнего сигнала внутрь клетки.
71. Циклический аденозинмонофосфат – строение, синтез, распад, роль в клетке. Факторы, влияющие на синтез и распад циклического аденозинмонофосфата.
(ответы совмещены)
Гормоны дистантного действия. К гормонам дистантного действия относятся гидрофильные (растворимые в воде) гормоны – катехоламины и гормоны белково-пептидной природы. Так как эти вещества не растворимы в липидах, они не могут проникать через клеточные мембраны. Рецепторы для этих гормонов расположены на наружной поверхности плазматической мембраны клеток-мишеней. Гормоны дистантного действия реализуют своё действие на клетку при помощи вторичного посредника, в качестве которого чаще всего выступает циклический АМФ (цАМФ).
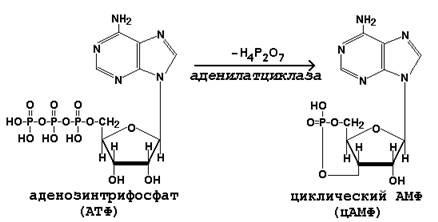
Циклический АМФ синтезируется из АТФ под действием аденилатциклазы:
Механизм дистантного действия гормонов показан на рисунке 23.3.
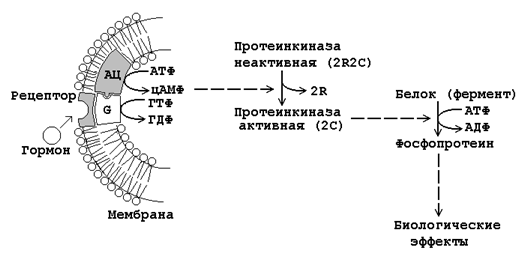
Рисунок 23.3. Механизм влияния на клетку гормонов дистантного действия.
Взаимодействие гормона с его специфическим рецептором приводит к активации G-белка клеточной мембраны. G-белок связывает ГТФ и активирует аденилатциклазу.
Активная аденилатциклаза превращает АТФ в цАМФ, цАМФ активирует протеинкиназу.
Неактивная протеинкиназа представляет собой тетрамер, который состоит из двух регуляторных (R) и двух каталитических (C) субъединиц. В результате взаимодействия с цАМФ происходит диссоциация тетрамера и освобождается активный центр фермента.
Протеинкиназа фосфорилирует белки-ферменты за счёт АТФ, либо активируя их, либо инактивируя. В результате этого изменяется (в одних случаях – увеличивается, в других – уменьшается) скорость химических реакций в клетках-мишенях.
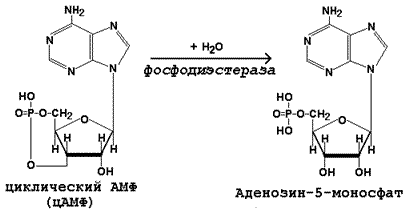
Инактивация цАМФ происходит при участии фермента фосфодиэстеразы:
72. Гормоны передней доли гипофиза - строение, механизм действия, биологическая роль. Последствия нарушений функции гипофиза в разные возрастные периоды.
Гормоны аденогипофиза. Аденогипофиз (передняя доля гипофиза) продуцирует и выделяет в кровь ряд тропных гормонов, регулирующих функцию как эндокринных, так и неэндокринных органов. Все гормоны гипофиза являются белками или пептидами. Внутриклеточным посредником всех гипофизарных гормонов (кроме соматотропина и пролактина) служит циклический АМФ (цАМФ). Характеристика гормонов передней доли гипофиза приводится в таблице 3.
| Гормон | Ткань-мишень | Основные биологические эффекты | Регуляция секреции |
|---|---|---|---|
| Адренокортикотропный гормон (АКТГ) | Кора надпочечников | Стимулирует синтез и секрецию стероидов корой надпочечников | Стимулируется кортиколиберином |
| Тиреотропный гормон (ТТГ) | Щитовидная железа | Усиливает синтез и секрецию тиреоидных гормонов | Стимулируется тиреолиберином и подавляется тиреоидными гормонами |
| Соматотропный гормон (гормон роста, СТГ) | Все ткани | Стимулирует синтез РНК и белка, рост тканей, транспорт глюкозы и аминокислот в клетки, липолиз | Стимулируется соматолиберином, подавляется соматостатином |
| Фолликулостимулирующий гормон (ФСГ) | Семенные канальцы у мужчин, фолликулы яичников у женщин | У мужчин повышает образование спермы, у женщин – образование фолликулов | Стимулируется люлиберином |
| Лютеинизирующий гормон (ЛГ) | Интерстициальные клетки семенников (у мужчин) и яичников (у женщин) | Вызывает секрецию эстрогенов, прогестерона у женщин, усиливает синтез и секрецию андрогенов у мужчин | Стимулируется люлиберином |
| Пролактин | Молочные железы (альвеолярные клетки) | Стимулирует синтез белков молока и развитие молочных желёз | Подавляется пролактостатином |
| Меланоцитостимулирующий гормон (МСГ) | Пигментные клетки | Повышает синтез меланина в меланоцитах (вызывает потемнение кожи) | Подавляется меланостатином |
73. Гормоны задней доли гипофиза: вазопрессин и окситоцин. Строение, механизм действия, биологическая роль. Последствия нарушения продукции вазопрессина.
Гормоны нейрогипофиза. К гормонам, секретируемым в кровоток задней долей гипофиза, относятся окситоцин и вазопрессин. Оба гормона синтезируются в гипоталамусе в виде белков-предшественников и перемещаются по нервным волокнам в заднюю долю гипофиза.
Окситоцин – нонапептид, вызывающий сокращения гладкой мускулатуры матки. Он используется в акушерстве для стимуляции родовой деятельности и лактации.
Вазопрессин – нонапептид, выделяемый в ответ на повышение осмотического давления крови. Клетками-мишенями для вазопрессина являются клетки почечных канальцев и гладкомышечные клетки сосудов. Действие гормона опосредовано цАМФ. Вазопрессин вызывает сужение сосудов и повышение артериального давления, а также усиливает реабсорбцию воды в почечных канальцах, что приводит к снижению диуреза.
23.4.4. Основные виды нарушений гормональной функции гипофиза и гипоталамуса. При дефиците соматотропного гормона, возникающем в детском возрасте, развивается карликовость (низкий рост). При избытке соматотропного гормона, возникающем в детском возрасте, развивается гигантизм (аномально высокий рост).
При избытке соматотропного гормона, возникающем у взрослых (в результате опухоли гипофиза), развивается акромегалия – усиленный рост кистей рук, ступней, нижней челюсти, носа.
При недостатке вазопрессина, возникающем вследствие нейротропных инфекций, черепно-мозговых травм, опухолей гипоталамуса, развивается несахарный диабет. Основным симптомом этого заболевания является полиурия – резкое увеличение диуреза при пониженной (1,001 – 1,005) относительной плотности мочи.
74. Инсулин - строение, образование из проинсулина, регуляция секреции инсулина, взаимодействие инсулина с рецептором.
75. Изменения активности внутриклеточных ферментов под действием инсулина, влияние инсулина на обмен веществ.
(ответ совмещен)
Инсулин. Инсулин – белково-пептидный гормон, вырабатываемый β-клетками островков Лангерганса. Молекула инсулина состоит из двух полипептидных цепей (А и В), содержащих 21 и 30 аминокислотных остатков соответственно; цепи инсулина связаны между собой двумя дисульфидными мостиками. Образуется инсулин из белка-предшественника (препроинсулина) путём частичного протеолиза (см. рисунок 4). После отщепления сигнальной последовательности образуется проинсулин. В результате ферментативного превращения удаляется фрагмент полипептидной цепи, содержащий около 30 аминокислотных остатков (С-пептид), и образуется инсулин.
Стимулом для секреции инсулина является гипергликемия – повышение содержания глюкозы в крови (например, после приёма пищи). Главные мишени для инсулина – клетки печени, мышц и жировой ткани. Механизм действия – дистантный.
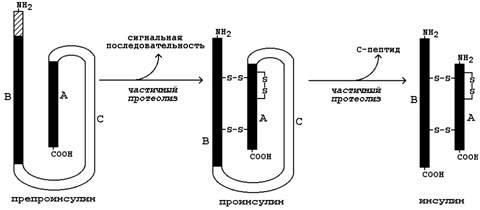
Рисунок 4. Схема превращения препроинсулина в инсулин.
Рецептор инсулина представляет собой сложный белок – гликопротеин, расположенный на поверхности клетки-мишени. Этот белок состоит их двух α-субъединиц и двух β-субъединиц, связанных между собой дисульфидными мостиками. β-Субъединицы содержат несколько аминокислотных остатков тирозина. Рецептор инсулина обладает тирозинкиназной активностью, т.е. способен катализировать перенос остатков фосфорной кислоты от АТФ на ОН-группу тирозина (рисунок 5).
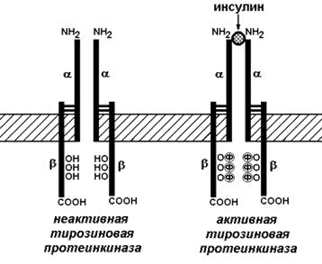
Рисунок 5. Инсулиновый рецептор.
В отсутствие инсулина рецептор не проявляет ферментативной активности. При связывании с инсулином рецептор подвергается аутофосфорилированию, т.е. β-субъединицы фосфорилируют друг друга. В результате изменяется конформация рецептора и он приобретает способность фосфорилировать другие внутриклеточные белки. В дальнейшем комплекс инсулина с рецептором погружается в цитоплазму и его компоненты расщепляются в лизосомах.
Образование гормон-рецепторного комплекса повышает проницаемость клеточных мембран для глюкозы и аминокислот. Под действием инсулина в клетках-мишенях:
а) снижается активность аденилатциклазы и увеличивается активность фосфодиэстеразы, что приводит к понижению концентрации цАМФ;
б) повышается скорость окисления глюкозы и снижается скорость глюконеогенеза;
в) увеличивается синтез гликогена и жиров и подавляется их мобилизация;
г) ускоряется синтез белка и тормозится его распад.
Все эти изменения направлены на ускоренное использование глюкозы, что приводит к снижению содержания глюкозы в крови. Инактивация инсулина происходит главным образом в печени и заключается в разрыве дисульфидных связей между цепями А и В.
76. Глюкагон - строение, факторы, влияющие на секрецию, механизм действия и биологическая роль
Глюкагон. Глюкагон – полипептид, содержащий 29 аминокислотных остатков. Он продуцируется α-клетками островков Лангерганса в виде белка-предшественнника (проглюкагона). Частичный протеолиз прогормона и секреция глюкагона в кровь происходит при гипогликемии, вызванной голоданием.
Клетки-мишени для глюкагона – печень, жировая ткань, миокард. Механизм действия – дистантный (посредником является цАМФ).
Под действием глюкагона в клетках-мишенях:
а) ускоряется мобилизация гликогена в печени (см. рисунок 6) и тормозится его синтез;
б) ускоряется мобилизация жиров (липолиз) в жировой ткани и тормозится их синтез;
в) угнетается синтез белка и усиливается его катаболизм;
г) ускоряется глюконеогенез и кетогенез в печени.
Конечный эффект глюкагона – поддержание высокого уровня глюкозы в крови.
77. Биохимические изменения при сахарном диабете. Метаболические механизмы развития осложнений при сахарном диабете. Последствия длительной гипергликемии. Особенности сахарного диабета у детей.
Согласно данным Всемирной организации здравоохранения, сахарный диабет классифицируют с учётом различия генетических факторов и клинического течения на две основные формы: диабет I типа - инсулинзависимый (ИЗСД), и диабет II типа - инсулиннезависимый (ИНСД).
1. Инсулинзависимый сахарный диабет
Инсулинзависимый сахарный диабет - заболевание, вызываемое разрушением р-клеток островков Лангерханса поджелудочной железы.
Деструкция β-клеток - результат аутоиммунных реакций. В аутоиммунной реакции принимают участие лимфоциты и макрофаги (моноциты). Эти клетки продуцируют цитокины, которые либо непосредственно повреждают β-клетки, либо опосредуют клеточные реакции против β-клеток.
Провоцировать возникновение диабета I типа может вирусная инфекция, вызывающая деструкцию b-клеток. К таким вирусам, называемым β-цитотропными, относят вирусы оспы, краснухи, кори, цитомегаловирус, эпидемического паротита, Коксаки, аденовирус. Некоторые р-цитотропные вирусы вызывают лизис β-клеток.
Известны некоторые токсические вещества, например, такие как производные нитрозомочевины и другие нитро- или аминосодержащие соединения, избирательно поражающие β-клетки и индуцирующие аутоиммунную реакцию. Кроме того, ИЗСД может быть результатом частичного генетически обусловленного дефекта системы иммунологического надзора и сочетаться с другими аутоиммунными заболеваниями. На долю ИЗСД приходится примерно 25-30% всех случаев сахарного диабета. Как правило, разрушение β-клеток происходит медленно, и начало заболевания не сопровождается нарушениями метаболизма. Когда погибает 80-95% клеток, возникает абсолютный дефицит инсулина, и развиваются тяжёлые метаболические нарушения. ИЗСД поражает в большинстве случаев детей, подростков и молодых людей, но может проявиться в любом возрасте (начиная с годовалого).
2. Инсулинонезависимый сахарный диабет
Инсулинонезависимый сахарный диабет - общее название нескольких заболеваний, развивающихся в результате относительного дефицита инсулина, возникающего вследствие нарушения секреции инсулина, нарушения превращения проинсулина в инсулин, повышения скорости катаболизма инсулина, а также повреждения механизмов передачи инсулинового сигнала в клетки-мишени (например, дефекта рецептора инсулина, повреждения внутриклеточных посредников инсулинового сигнала и др.). ИНСД поражает людей, как правило, старше 40 лет. Сахарный диабет II типа характеризуется высокой частотой семейных форм. Риск ИНСД у ближайших родственников больного достигает 50%, тогда как при ИЗСД он не превышает 10%. Заболевание поражает преимущественно жителей развитых стран, особенно горожан.
Возможными причинами ИНСД могут быть: образование антител к рецепторам инсулина; генетический дефект пострецепторного аппарата инсулинзависимых тканей; нарушения регуляции секреции инсулина. К факторам, определяющим развитие и клиническое течение болезни, относят ожирение, неправильный режим питания, малоподвижный образ жизни, стресс.
Мутации генов, контролирующих секрецию инсулина, энергетический обмен в β-клетках и обмен глюкозы в клетках-мишенях инсулина, приводят к возникновению нескольких форм ИНСД с аутосомно-доминантным наследованием.
Основным провоцирующим фактором инсулинонезависимого диабета служит ожирение.
Этот тип диабета часто сочетается с гиперинсулинемией, что способствует ожирению. Таким образом, ожирение, с одной стороны, важнейший фактор риска, а с другой - одно из ранних проявлений сахарного диабета.
При сахарном диабете, как правило, соотношение инсулин/глюкагон снижено. При этом ослабевает стимуляция процессов депонирования гликогена и жиров, и усиливается мобилизация запасов энергоносителей. Печень, мышцы и жировая ткань даже после приёма пищи функционируют в режиме постабсорбтивного состояния.
1. Симптомы сахарного диабета
Для всех форм диабета характерно повышение концентрации глюкозы в крови - гипергликемия.После приёма пищи концентрация глюкозы может достигать 300-500 мг/дл и сохраняется на высоком уровне в постабсорбтивном периоде, т.е. снижается толерантность к глюкозе. Снижение толерантности к глюкозе наблюдают в случаях скрытой (латентной) формы

Рис. 11-30. Изменение толерантности к глюкозе у больных скрытой формой сахарного диабета. Определение толерантности к глюкозе используют для диагностики сахарного диабета. Обследуемый принимает раствор глюкозы из расчёта 1 г на 1 кг массы тела (сахарная нагрузка). Концентрацию глюкозы в крови измеряют в течение 2-3 ч с интервалами в 30 мин. 1 - у здорового человека; 2 - у больного сахарным диабетом.
сахарного диабета. В этих случаях у людей отсутствуют жалобы и клинические симптомы, характерные для сахарного диабета, а концентрация глюкозы в крови натощак соответствует норме. Однако использование провокационных проб (например, сахарной нагрузки) выявляет снижение толерантности к глюкозе (рис. 11-30).
Повышение концентрации глюкозы в плазме крови обусловлено снижением скорости использования глюкозы тканями вследствие недостатка инсулина или снижения биологического действия инсулина в тканях-мишенях.
При дефиците инсулина уменьшается количество белков-переносчиков глюкозы (ГЛЮТ-4) на мембранах инсулинзависимых клеток (жировой ткани и мышц). В мышцах и печени глюкоза не депонируется в виде гликогена, в жировой ткани уменьшается скорость синтеза и депонирования жиров. Кроме того, при снижении инсулинглюкагонового индекса активируется глюконеогенез из аминокислот, глицерола и лактата. Повышение концентрации глюкозы в крови при сахарном диабете превышает концентрационный почечный порог, что становится причиной выделения глюкозы с мочой (глюкозурия). В норме проксимальные канальцы почек реабсорбируют всю фильтрующуюся в клубочках глюкозу, если её уровень не превышает 8,9 ммоль/л (160 мг/дл).
К характерным признакам сахарного диабета относят также повышение концентрации в крови кетоновых тел - кетонемия. При низком соотношении инсулин/глюкагон жиры не депонируются, а ускоряется их катаболизм, так как гормончувствительная липаза в жировой ткани находится в фосфорилированной активной форме. Концентрация неэтерифицирован-ных жирных кислот в крови повышается. Печень захватывает жирные кислоты, окисляет их до ацетил-КоА, который, в свою очередь, превращается в β-гидроксимасляную и ацетоуксусную кислоты. В тканях ацетоацетат частично декарбоксилируется до ацетона, запах которого исходит от больных сахарным диабетом и ощущается даже на расстоянии. Увеличение концентрации кетоновых тел в крови (выше 20 мг/дл, иногда до 100 мг/дл) приводит к кетонурии. Накопление кетоновых тел снижает буферную ёмкость крови и вызывает ацидоз.
593
Ещё один характерный признак сахарного диабета - повышенный уровень в крови ли-попротеинов (в основном, ЛПОНП) - гипер-липопротеинемия. Пищевые жиры не депонируются в жировой ткани вследствие ослабления процессов запасания, а поступают в печень, где частично превращаются в триацилглицеролы, которые транспортируются из печени в составе ЛПОНП.
При сахарном диабете дефицит инсулина приводит к снижению скорости синтеза белков в организме и усилению распада белков. Это вызывает повышение концентрации аминокислот в крови. Аминокислоты поступают в печень и дезаминируются. Безазотистые остатки гликогенных аминокислот включаются в глюконеогенез, что ещё более усиливает гипергликемию. Образующийся при этом аммиак вступает в орнитиновый цикл, что приводит к увеличению концентрации мочевины в крови и, соответственно, в моче - азотемия и азотурия.
Высокие концентрации глюкозы, кетоновых тел, мочевины требуют усиленной экскреции их из организма. Поскольку концентрационная способность почек ограничена, резко увеличивается выделение большого количества воды, в результате чего может наступить обезвоживание организма. Выделение мочи у больных возрастает в несколько раз и в некоторых случаях достигает 8-9 л в сутки, но чаще не превышает 3-4 л - полиурия. Потеря воды вызывает постоянную жажду - полидипсия.
2. Острые осложнения сахарного диабета.
Механизмы развития диабетической комы
Нарушения обмена углеводов, жиров и белков при сахарном диабете могут приводить к развитию коматозных состояний (острые осложнения). Диабетическая кома проявляется в резком нарушении всех функций организма с потерей сознания. Основные предшественники диабетической комы - ацидоз и дегидратация тканей (рис. 11-31).
Параллельно кетоацидозу при декомпенсации диабета развивается нарушение водно-электролитного обмена. В его основе лежит гипергликемия, сопровождающаяся повышением осмотического давления в сосудистом русле. Для сохранения осмолярности начинается компенсаторное перемещение жидкости из клеток и внеклеточного пространства в сосудистое русло. Это ведёт к потере тканями воды и электролитов, прежде всего ионов Na+, K+, С1-, НСО3. В результате развиваются тяжёлая клеточная дегидратация и дефицит внутриклеточных ионов (прежде всего К+), затем возникает общая дегидратация. Это приводит к снижению периферического кровообращения, уменьшению мозгового и почечного кровотока и гипоксии. Диабетическая кома развивается медленно, в течение нескольких дней, но иногда может возникнуть и в течение нескольких часов. Первыми признаками могут быть тошнота, рвота, заторможенность. АД у больных снижено.
Коматозные состояния при сахарном диабете могут проявляться в трёх основных формах: кетоацидотической, гиперосмолярной и лакто-ацидотической. Для кетоацидотической комы характерны выраженный дефицит инсулина, кетоацидоз, полиурия, полидипсия. Гипергликемия (20-30 ммоль/л), обусловленная инсулиновой недостаточностью, сопровождается большими потерями жидкости и электролитов, дегидратацией и гиперосмоляльностью плазмы. Общая концентрация кетоновых тел достигает 100 мг/дл и выше.
При гиперосмолярной коме наблюдают чрезвычайно высокие уровни глюкозы в плазме крови, полиурию, полидипсию, всегда проявляется тяжёлая дегидратация. Предполагают, что у большинства больных гипергликемия обусловлена сопутствующим нарушением функции почек. Кетоновые тела в сыворотке крови обычно не определяются.
При лактоацидотической коме преобладают гипотония, снижение периферического кровообращения, гипоксия тканей, приводящая к смещению метаболизма в сторону анаэробного гликолиза, что обусловливает повышение концентрации молочной кислоты в крови (лакто-ацидоз).
Разные варианты диабетической комы в чистом виде практически не встречаются. Их возникновение может быть обусловлено разными факторами, например инфекционными заболеваниями, травмами, хирургическими вмешательствами, токсическими соединениями и др.
3. Поздние осложнения сахарного диабета
Главная причина поздних осложнений сахарного диабета - гипергликемия. Гипергликемия приводит к повреждению кровеносных сосудов

Рис. 11-31. Изменение метаболизма при сахарном диабете и причины диабетической комы.
595
и нарушению функций различных тканей и органов.
Одним из основных механизмов повреждения тканей при сахарном диабете являетсягликозилирование белков, приводящее к изменению их конформации и функций. Некоторые белки в норме содержат углеводные компоненты, причём образование таких гликопротеинов протекает ферментативно (например, образование гликопротеиновых гормонов аденогипофиза). Однако в организме человека может происходить и неферментативное взаимодействие глюкозы со свободными аминогруппами белков - неферментативное гликозилирование белков. В тканях здоровых людей эта реакция протекает медленно. При гипергликемии процесс гликозилирования ускоряется. Степень гликозилирования белков зависит от скорости их обновления. В медленно обменивающихся белках накапливается больше изменений. К одним из первых признаков сахарного диабета относят увеличение в 2-3 раза количества гликозилированного гемоглобина (норма НbА1С5,8-7,2%). Другим примером медленно обменивающихся белков служат кристаллины - белки хрусталика. При гликозилировании кристаллины образуют многомолекулярные агрегаты, увеличивающие преломляющую способность хрусталика. Прозрачность хрусталика уменьшается, возникает его помутнение, или катаракта.
К медленно обменивающимся белкам относятся белки межклеточного матрикса, базальных мембран. Утолщение базальных мембран, одно из характерных осложнений сахарного диабета, приводит к развитию диабетических ангиопатий.
Причиной многих поздних осложнений сахарного диабета также служит повышение скорости превращения глюкозы в сорбитол (см. раздел 7).
- Реакция превращения глюкозы в шестиатомный спирт (сорбитол) катализируется ферментом альдозоредуктазой. Сорбитол не используется в других метаболических путях, а скорость его диффузии из клеток невелика. У больных сахарным диабетом сорбитол накапливается в сетчатке и хрусталике глаза, клетках клубочков почек, шванновских клетках, в эндотелии.
- Сорбитол в высоких концентрациях токсичен для клеток. Его накопление в нейронах приводит к увеличению осмотического давления, набуханию клеток и отёку тканей. Так, например, помутнение хрусталика может развиться вследствие вызванного накоплением сорбитола набухания хрусталика и нарушения упорядоченной структуры кристаллинов.
Диабетические ангиопатий. Диабетические ангиопатий обусловлены прежде всего поражением базальных мембран сосудов. При высокой концентрации глюкозы в плазме крови протеогликаны, коллагены, гликопротеины гликозилируются, нарушается обмен и соотношение между компонентами базальных мембран, нарушается их структурная организация.
- Макроангиопатии проявляются в поражениях крупных и средних сосудов сердца, мозга, нижних конечностей. Патологические изменения во внутренней оболочке артерий и повреждения артериальной стенки в средних и наружных слоях - следствие гликозилирования базальных мембран и белков межклеточного матрикса (коллагена и эластина), что приводит к снижению эластичности артерий. В сочетании с гиперли-пидемией это может быть причиной развития атеросклероза. При сахарном диабете атеросклероз встречается чаще, развивается в более раннем возрасте и прогрессирует значительно быстрее, чем в отсутствие диабета.
- Микроангиопатии - результат повреждения капилляров и мелких сосудов. Проявляются в форме нефро-, нейро- и ретинопатии.
Нефропатия развивается примерно у трети больных сахарным диабетом. Электронно-микроскопические изменения базальной мембраны в почечных клубочках можно обнаружить уже на первом году после установления диагноза. Однако у большинства больных клинические признаки диабетической нефропатии проявляются через 10-15 лет существования диабета. Признаком ранних стадий нефропатии служит микроальбуминурия (в пределах 30-300 мг/сут), которая в дальнейшем развивается до классического нефротического синдрома, характеризующегося высокой протеинурией, гипоальбуминемией и отёками.
Ретинопатия, самое серьёзное осложнение сахарного диабета и наиболее частая причина слепоты, развивается у 60-80% больных сахарным
596
диабетом. На ранних стадиях развивается базальная ретинопатия, которая проявляется в кровоизлияниях в сетчатку, расширении сосудов сетчатки, отёках, Если изменения не затрагивают жёлтого пятна, потеря зрения обычно не происходит. В дальнейшем может развиться пролиферативная ретинопатия, проявляющаяся в новообразовании сосудов сетчатки и стекловидного тела. Ломкость и высокая проницаемость новообразованных сосудов определяют частые кровоизлияния в сетчатку или стекловидное тело. На месте тромбов развивается фиброз, приводящий к отслойке сетчатки и потере зрения.
78. Адреналин - механизм действия и биологическая роль, строение, реакции образования адреналина из тирозина.
К гормонам мозгового вещества надпочечников относятся адреналин и норадреналин (катехоламины). Они синтезируются в хромаффинных клетках из тирозина (рисунок 7).
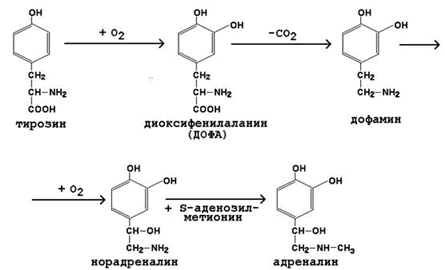
Рисунок 7. Схема синтеза катехоламинов.
Секреция адреналина усиливается при стрессе, физических нагрузках. Мишени для катехоламинов – клетки печени, мышечной и жировой ткани, сердечно-сосудистая система. Механизм действия – дистантный. Эффекты реализуются через аденилатциклазную систему и проявляются изменениями углеводного обмена. Подобно глюкагону, адреналин вызывает активацию мобилизации гликогена (см. рисунок 6) в мышцах и печени, липолиз в жировой ткани. Это приводит к увеличению содержания глюкозы, лактата и жирных кислот в крови. Адреналин усиливает также сердечную деятельность, вызывает сужение сосудов.
Обезвреживание адреналина происходит в печени. Основными путями обезвреживания являются: метилирование (фермент – катехол-орто-метилтрансфераза, КОМТ), окислительное дезаминирование (фермент – моноаминооксидаза, МАО) и конъюгация с глюкуроновой кислотой. Продукты обезвреживания выводятся с мочой.
79. Глюкокортикоиды – образование, механизм действия, биологическая роль, строение. Метаболические изменения при избытке глюкокортикоидов.
Глюкокортикоиды. К ним относятся кортизол (другое название - гидрокортизон), кортикостерон, кортизон. Это стероидные гормоны, синтезируются они на основе холестерола. Синтез глюкокортикоидов регулируется адренокортикотропным гормоном (АКТГ) гипофиза (см. таблицу 2). Секреция глюкокортикоидов усиливается при стрессе. Для этих гормонов характерен прямой механизм действия: гормон → ген → мРНК → белок (фермент). Ткани-мишени: мышцы, жировая и лимфоидная ткани, печень, почки.
Запомните основные эффекты глюкокортикоидов:
а) в мышечной и лимфоидной тканях глюкокортикоиды ингибируют синтез белков и усиливают их распад. Это вызывает поступление большого количества свободных аминокислот в кровь;
б) в печени и почках глюкокортикоиды усиливают синтез многих белков, в том числе аминотрансфераз и ферментов глюконеогенеза. Это благоприятствует использованию свободных аминокислот для синтеза глюкозы. Синтезированная глюкоза поступает в кровь; частично она используется для синтеза гликогена в печени и мышцах;
в) глюкокортикоиды усиливают мобилизацию (расщепление) жиров в жировой ткани; образующийся глицерол поступает в печень и включается в глюконеогенез; жирные кислоты подвергаются окислению, продукты которого используются в синтезе кетоновых тел.
80. Минералокортикоиды – механизм действия, биологическая роль, строение. Метаболические изменения при избытке и недостатке минералокортикоидов.
Минералокортикоиды. Представители этой группы - альдостерон (см. рисунок), дезоксикортикостерон - также являются стероидными гормонами и образуются из холестерола. Синтез минералокортикоидов регулируется АКТГ и ангиотензином II (пептидом, образующимся из белка плазмы крови ангиотензиногена путём частичного протеолиза). Минералокортикоиды - гормоны прямого действия, мишенями служат клетки эпителия дистальных канальцев почек. Под действием альдостерона в клетках-мишенях активируется синтез белков, участвующих в транспорте Na+ через клеточные мембраны эпителия канальцев. В результате усиливается реабсорбция Na+ и Cl- из мочи в межклеточную жидкость и далее в кровь. Вместе с Na+ пассивно следует вода. Одновременно в мочу выделяются ионы К+ (в обмен на Na+) Таким образом, альдостерон способствует задержке в тканях Na+ и воды и потере с мочой К+. Инактивация глюко- и минералокортикоидов происходит в печени, конечными продуктами являются 17-кетостероиды, которые выводятся с мочой.
29.2.3. Нарушения гормональной функции надпочечников. Основные проявления гипер- и гипофункции коры надпочечников представлены в таблице 4.
| Показатели | Гиперфункция коры надпочечников (гиперкортицизм, болезнь Иценко-Кушинга) |
Гипофункция коры надпочечников (гипокортицизм, болезнь Аддисона) |
|---|---|---|
| Этиология заболевания | Развивается при опухоли надпочечника, а также при опухоли гипофиза с повышенной продукцией АКТГ. | Возникает в результате туберкулёзного поражения надпочечников либо вследствие пониженной секреции АКТГ. |
|
Основные симптомы |
Ожирение - скопление жира в области лица и туловища; отёки; повышение артериального давления; остеопороз - пустоты в костях, вызванные нарушением синтеза коллагена и деминерализацией; стероидный диабет. | У больных снижена устойчивость к эмоциональному стрессу, инфекциям, травмам. Артериальное давление снижено, мышечная слабость, быстрая утомляемость. Больные погибают из-за нарушений водно-солевого баланса. |
|
Изменения состава крови |
Повышение содержания глюкозы, мочевины, аминокислот, жирных кислот, кетоновых тел, ионов натрия, снижение содержания ионов калия в крови | Снижение содержания глюкозы, мочевины, аминокислот, жирных кислот, кетоновых тел, ионов натрия, повышение содержания ионов калия в крови |
|
Изменения состава мочи |
Повышение экскреции аминокислот, мочевины, ионов калия, снижение экскреции ионов натрия, появление в моче глюкозы и кетоновых тел, снижение диуреза | Снижение экскреции аминокислот, мочевины, ионов калия, повышение экскреции ионов натрия, увеличение диуреза |
ОБНОВЛЕНИЯ
08 июня 2019, 16:18:59
08 июня 2019, 16:10:24
07 июня 2019, 22:59:05
09 мая 2019, 16:13:23
27 апреля 2019, 16:00:00
24 апреля 2019, 01:02:18
17 апреля 2019, 22:26:23
17 апреля 2019, 22:21:37
17 апреля 2019, 22:07:42
17 апреля 2019, 22:06:28
ПОДПИСАТЬСЯ НА РАССЫЛКУ
ПРЕДМЕТЫ
- Анатомия
- Акушерство и гинекология
- БЖД, медицина катастроф
- Биохимия
- Биология
- Гистология
- Гигиена
- Генетика
- Диетология
- Дерматовенерология
- Инфекционные болезни
- Культурология
- Лабораторная диагностика
- Летняя практика
- Лучевая диагностика
- Медицинская информатика
- Микробиология
- Неврология
- Общественное здоровье
- Общий уход
О НАС
«Dendrit» - информационный портал для медицинских работников, студентов медицинских ВУЗов, исследователей и пациентов.
Ваш источник новостей и знаний о здоровье.
dendrit.ru © 2014-2024
